By Ioannis
Psarommatis MD, PhD
Introduction
Auditory Neuropathy: the torment of modern researchers and
audiologists and a real challenge to their diagnostic abilities and their understanding
of the auditory function.
At the same time, auditory
neuropathy might prove to be an excellent chance for breaking away from the
standard audiological traditions and bridge together many scientific fields.
It is very possible that the extremely familiar term sensorineural hearing
loss will NOT have precise meaning in the near future.
Clinical approach to auditory neuropathy
During the last few years, Auditory Neuropathy
(AN) has been established as a well-accepted clinical entity. The term is ascribed
to Starr
et al [1]who 10 years ago published
the first report on AN. The data he discussed were based on 10 patients
having: (i) mild to severe hearing loss ; (ii) poor speech discrimination scores;
(iii) absent or severely disturbed auditory brainstem responses (ABR); (iv)
normal otoacoustic emissions (OAEs);and (v)
no central nervous system abnormalities . They have attributed their findings
to a functional disorder of the acoustic nerve. In this context it should
be noted that Auditory Neuropathy-like cases had been reported
several years earlier [2, 3, 4]
The last decade the introduction of OAEs in the everyday clinical practice
has resulted in diagnosing AN at increasing rates. It is not surprising, therefore,
that numerous reports have appeared recently in the international literature.
Today we use the definition for auditory neuropathy patients, whose clinical
and audiological characteristics are summarized in Table
1.
Table 1*: Typical clinical and laboratory findings in patients with auditory
neuropathy |
- Variable pure tone audiometry (from normal hearing to severe/profound
hearing loss). Any configuration possible. Uni- or bilateral.
- Poor speech recognition scores (worse than that predicted from PTA, particularly
in noise).
- Otoacoustic emissions (TEOAEs or DPOAEs) present.
- Click-evoked auditory brainstem responses absent or severely abnormal.
- Cochlear microphonic present.
- Acoustic stapedious muscle reflexes (ipsi- and contralateral) absent.
- Suppression of TEOAEs by contralateral noise absent.
- Masking level difference not detectable.
- Gap detection absent.
- No central nervous system abnormalities on MRI to account for these findings.
|
| * Modified from Linda J. Hood, Charles I. Berlin, Thierry Morlet, Shanda
Brashears, Kelly Rose, Sonya Tedesco. Considerations in the Clinical Evaluation
of Auditory Neuropathy/Auditory Dys-Synchrony. Seminars in Hearing (2002)
23 (3): 201-7. |
However, there
is no consensus about the term Auditory Neuropathy;. Neural synchrony
disorder, brainstem auditory processing syndrome, central auditory dysfunction,
auditory synaptopathy, auditory dys-synchrony and primary auditory neuropathy
have also been used to describe patients showing similar audiological findinds.
The prevalence of the recently described pathology
known as ‘‘auditory neuropathy’’ in children with hearing loss has been reported
to vary greatly, ranging between 0.5 and 24%
[4, 5, 6, 7,
8, 9 ]
while the prevalence of auditory neuropathy in the non-risk population is unknown.
It is estimated that one in every ten children with hearing loss may suffer from
auditory neuropathy [
10 ].
Concerning the etiology of auditory neuropathy,
it is not well understood yet. Theoretically, all pathologies which affect
transmission of the auditory signal from the level of inner hair cells to auditory
cortex could give clinical and physiological evidences of what we today call “auditory
neuropathy”.
In such cases, if the necessary imaging studies fail to reveal a space occupying
lesion or anatomic abnormalities [
11] we can conclude
that one or more intrinsic factors of the auditory pathway could account for
these findings. Therefore, regardless the underlying pathology, auditory neuropathy
may be considered as an intrinsic dysfunction of the auditory pathways. Patients
meeting the profile of auditory neuropathy may be phenotypically healthy or
they may suffer from various and seemingly dissimilar pathologies, including
neonatal jaundice and anoxia, infections, neurologic, endocrine, metabolic
and genetic diseases
(Table 2> from references 9,12, 13, 14, 15, 16).
We can speculate that their harmful effect may be either:
a) Segmental or localized, limited
to a certain part of the auditory pathway (eg, otoferlin mutations that result
in defective exocytosis of neurotransmitter at the synapses between inner hair
cells and auditory nerve [17])
b) Multilevel or generalized, affecting a
great part of the auditory pathway (eg, bilirubin-induced damages on auditory
nuclei and probably auditory nerve [18, 19,]).
Table 2: Medical conditions associated with
Auditory Neuropathy
- Neonatal anoxia
- Premature birth
- Neonatal hyperbilirubinemia
- Infectious process (e.g., mumps, meningitis)
- Immune disorder (e.g., Guillain-Barré syndrome)
- Nonspecific febrile illness
- Hereditary sensory-motor neuropathy
(or Charcot-Marie-Tooth disease)
- Mitochondrial enzymatic defects
- Olivo-Pontine cerebellar degeneration
- Friedreich's ataxia
- Stevens-Johnson syndrome
- Ehlers-Danlos syndrome
- Spinocerebellar degeneration
- Leukodystrophy
- Gonad dysgenesis
- Seizures
- Leber's hereditary optic neuropathy
- Waardenburg’s syndrome
- Maple syrup urine disease
According to the transmission patterns, auditory
neuropathy could also be divided in a) genetic and b) non-genetic or acquired.
Neonates suffering from hyperbilirubinemia and anoxia may develop acquired
auditory neuropathy. On the contrary, mutations in the different chromosomes
and genes have been reported to be the cause of autosomal dominant or recessive
auditory neuropathy, either non-syndromic or accompanied with other inherited
neuropathies [
20, 21, 22, 23]. Recently, genetic
studies [
17, 24,25, 26] showed that mutations in
otoferlin gene cause non-syndromic recessive auditory neuropathy by affecting
calcium-mediated synaptic exocytosis of inner hair cells. Failure of neurotransmitter
release blocks the auditory signals at the level of the synapses between inner
hair cells and VIIIth nerve endings, thus resulting in a true “auditory synaptopathy”.
Another study [
27] has demonstrated that mutations in the gene encoding pejvakin,
a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory
neuropathy. Since this protein is expressed not only in the cochlear sensory
epithelium but in the cochlear nuclei, superior olivary complex and inferior
colliculus as well, it is believed that it may have a role in the propagation
of action potentials through auditory pathway and, consequently, its deficiency
could be responsible for an auditory neuropathy phenotype.
Prognosis of auditory neuropathy
remains unpredictable. There are variable reports in the literature showing
clinical or laboratory improvement of some patients [
6,
28, 29] while others
remain stable over time or even worsen, losing outer hair cells’ function
[5,
30, 31].
Moreover, some patients may intermittently suffer from auditory neuropathy
[
32]. In a large series of high risk infants we found that most infants meeting
the auditory neuropathy profile (13 out of 20, or 65%) demonstrated a restoration
of their auditory neural transmission, that is normal ABR recordings on re-examination.
Remarkably, recovery was significantly more common in infants with low birth
weight
[see reference
9 for details].
In any case, it becomes obvious that some subjects suffering from auditory
neuropathy will get better but we are not able to accurately predict who will
restore their auditory performance.
The remarkable variation in the clinical and laboratory findings and the unpredictable
prognosis observed in many auditory neuropathic patients makes their management
difficult, without commonly acceptable “rules”. Audiologists and otologists should avoid a strict approach to this unexplained pathology. On the contrary, a flexible attitude must be adopted taking into account individual needs. It is author’s
belief that in infants and young children behavioural tests should mainly direct
the management of auditory neuropathy, not the results of physiologic tests.
Hearing aids and cochlear implants are among treatment modalities but not all
auditory neuropathy cases have the indication or would benefit from them.
Studies have showed that
only some of the patients suffering from auditory neuropathy would benefit
from hearing aids [
5 , 33 ]. Probably, these
patients can hear but cannot understand [
34]. If
auditory neuropathy is purely an expression of de-synchronization of acoustic
nerve signalling, then hearing aids do not represent the management of choice.
Similar uncertainties and queries are encountered in cases treated with cochlear
implantation. Some researchers have reported favourable outcomes while others
have described poor speech perception scores in patients with auditory neuropathy
who have received a cochlear implant [
e.g.,
5,35, 36, 37 ]. The site and severity of lesion (-s) and the underlying
pathology may play a role for the observed diversity in surgical management
of auditory neuropathy. It is also possible electric stimuli provided by the
cochlear implant to re-synchronize auditory signalling, operating as a pacemaker
for the auditory pathway.
Hearing screening programs and auditory neuropathy
Since OAEs are generated within the cochlea,
OAE-based hearing screening can not detect neural dysfunction. Consequently,
infants with auditory neuropathy or neural conduction disorders without concomitant
sensory (ie, outer hair cell) dysfunction will not be detected by OAEs [38].
In such cases OAEs will give false negative results. Babies treated in neonatal
intensive care units are at increased risk for hearing loss, neural conduction
and/or auditory brainstem dysfunction, including auditory neuropathy [38].
Probably OAEs do not represent the method of choice for screening high risk populations.
The same could apply to all infants suffering from neurologic, metabolic, endocrine
or central nervous system disorder, where a higher rate of central auditory dysfunction
is expected. The impact of similar findings -that is the combination present
OAEs/abnormal ABR- on neonatal hearing screening programs will probably lead
to crucial changes in their schemes. Auditory brainstem responses (either automated
or conventional) supplemented by OAEs in cases showing elevated thresholds, atypical
or absent responses, may prove more effective methodology for screening high
risk neonates.
Case Studies
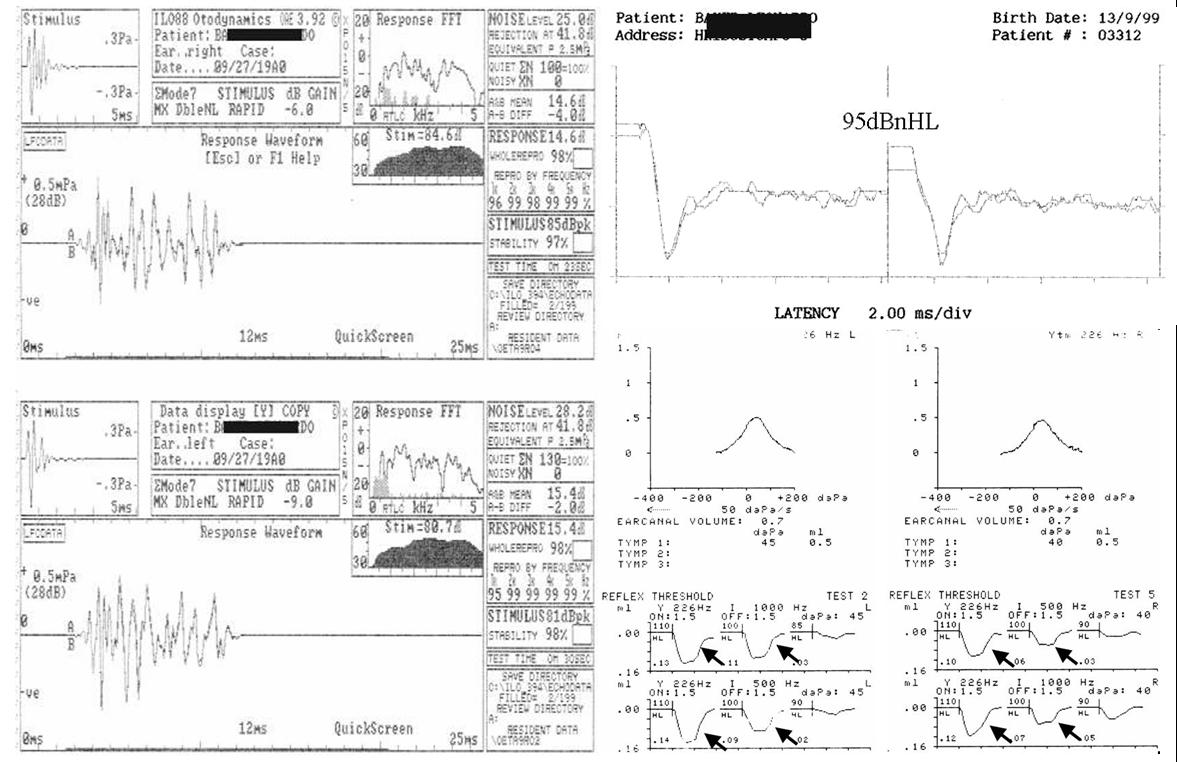
Case 1: Auditory Neuropathy with present acoustic middle
ear reflex.
Comments: In patients suffering from auditory neuropathy
acoustic middle ear reflex is typically absent. In this case, present OAEs
(left) and absent ABR (top-right) in a phenotypically normal child 1 ½ years
old are accompanied by present ipsilateral middle ear reflexes, bilaterally
(bottom-right, arrows).The Acoustic stapedious muscle reflex can be present
in patients suffering from auditory neuropathy. Tone-evoked stapedious reflex
may not need the strict synchronization required for ABR recordings.
Case 2: Auditory neuropathy in a child with pituitary stalk
interruption syndrome (PSIS)
Comments: Another unusual case of auditory neuropathy. A boy 2 years old was referred for audiologic evaluation due to a mild speech delay (first true words at the age of 15 months), mild hypotonia, clumsy walking and short stature. Auditory brainstem responses were consistent with severe to profound hearing loss bilaterally (top-left), TEOAEs were normal in both ears (top-right) while free-field audiometry suggested only mild hearing loss, if any. We did not recommend hearing amplification. At the same age (2y.o.) primary hypothyroidism was diagnosed. Physiologic tests were repeated at age 3 ½ yielding similar findings. Subjective tests revealed normal pure tone thresholds bilaterally (bottom-left) and near normal speech discrimination scores. Due to persistent short stature a detailed endocrine investigation was undertaken and growth hormone insufficiency was diagnosed. Pituitary MRI (bottom-right) was characteristic of pituitary stalk interruption with a hypoplastic anterior lobe of hypophysis (arrow) and a bright ectopic posterior lobe, just below the hypothalamus (arrowhead). The pituitary stalk is not visible. Today the child is 9 years old, he is under replacement therapy, he has never used hearing aids and he shows normal speech skills.
The combination of PSIS and auditory neuropathy represents a poorly understood pathology of unknown pathogenetic mechanism. It could be attributed to genetic interactions during the fetal growth, between genes which are involved in the embryogenesis of hypophysis and those responsible for auditory neuropathy phenotype.
Conclusions
Genetic or acquired, syndromic or non-syndromic, segmental or multilevel, permanent, transient or fluctuant, auditory neuropathy is an intrinsic dysfunction of auditory pathways with many unexplored aspects. We must first understand the underlying pathogenetic mechanism (-s) before drawing definitive conclusions about its terminology, etiology (-ies), prognosis and optimal management. Till then, the use of the controversial term “auditory neuropathy” probably
represents the less offensive action among those we have taken for this disorder.
Key issues relative to AN
• Interpret a positive screening OAE-test cautiously, especially in neonates from ICUs
• Repeat physiologic tests in newborns diagnosed with auditory neuropathy at about 6 months of age
• Inform/educate parents adequately
• Individualize treatment plan
• Use behavioural tests for decision making concerning hearing amplification and cochlear implantation
• If hearing aids are recommended, consider monaural amplification, low maximum power output, close monitoring with OAEs
• If cochlear implantation is recommended, allow adequate time to elapse because CNS maturation or auditory function restoration may be in progress
• Be well-informed about the latest developments on the subject
References
1. A. Starr, T. Pincton, Y. Sininger, L. Hood, C. Berlin. Auditory neuropathy. Brain 119 (1996) 741-753.
H. Davis, S.K. Hirsh. A slow brain stem response for low frequency audiometry. Audiology 18 (1979) 445-461.
2. H. Davis, S.K. Hirsh. A slow brain stem response for low frequency audiometry. Audiology 18 (1979) 445-461
3. D. Worthington, J. Peters, Quantifiable hearing and no ABR: paradox or error? Ear Hear. (1980) 1:281-285.
4. N. Kraus, O. Ozdamar, L. Stein, N. Reed. Absent auditory brain stem response: peripheral hearing loss or brain stem dysfunction? Laryngoscope 94 (1984) 400-406.
5. G. Rance, D. Beer, B. Cone-Wesson, R. Shepherd, R. Dowell, A. King, F. Rickards, G. Clark. Clinical findings for a group of infants and young children with auditory neuropathy. Ear Hear. 20 (1999) 238-252.
6. C. Madden, M. Rutter, L. Hilbert, J. Greinwald, D. Choo. Clinical and audiological features in auditory neuropathy. Arch. Otolaryngol. Head Neck. Surg. 128 (2002) 1026-1030.
7. C.I. Berlin, T. Morlet, L.J. Hood. Auditory neuropathy/dyssynchrony: its diagnosis and management. Pediatr. Clin. North Am. 50 (2003) 331-340.
8. A.L. Berg, J.B. Spitzer, H.M. Towers, C. Bartosiewicz, B.E. Diamond. Newborn hearing screening in the NICU: profile of failed auditory brainstem response/passed otoacoustic emission. Pediatrics 116 (4) (2005) 933-938.
9. Ioannis Psarommatis, Maria Riga, Konstantinos Douros, Petros Koltsidopoulos, Dimitrios Douniadakis, Ioannis Kapetanakis, Nikolaos Apostolopoulos. Transient infantile auditory neuropathy and its clinical implications. Int J Pediatr Otorhinolaryngol 70 (2006) 1629-1637.
10. Yvonne S. Sininger. Identification of Auditory Neuropathy in Infants and Children. Seminars in Hearing (2002) 23(3): 193-200.
11. Craig A. Buchman, Patricia A. Roush, Holly F. B. Teagle, Carolyn J. Brown, Carlton J. Zdanski, John H Grose. Auditory Neuropathy Characteristics in Children with Cochlear Nerve Deficiency. Ear & Hearing (2006) 27 (4): 399-408.
12. R. Tibesar, J.K. Shallop. Auditory neurapathy, in: C. Cummings, P. Flint, B. Haughey, T. Robbins, R. Thomas, L. Harker, M. Richardson, D. Schuller (Eds.), Otolaryngology: Head & Neck Surgery, 4th ed., Elsevier Mosby, Philadelphia, 2005, 3503-3521.
13. Y. Sininger, S. Oba. Patients with auditory neuropathy: who are they and what can they hear? in: Y. Sininger, A. Starr (Eds.), Auditory Neuropathy: A New Perspective on Hearing Disorders, Singular Press, San Diego, 2001, 15-35.
14: A. Starr. The neurology of auditory neuropathy, in: Y. Sininger, A. Starr (Eds.), Auditory Neuropathy: A New Perspective on Hearing Disorders, Singular Press, San Diego, 2001, 37-50.
15. Ceranic B, Luxon LM. Progressive auditory neuropathy in patients with Leber's hereditary optic neuropathy. J Neurol Neurosurg Psychiatry (2004) 75(4):626-30.
16. Beno?t Jutrasa, Laura J. Russell, Anne-Marie Hurteau, Martine Chapdelaine. Auditory neuropathy in siblings with Waardenburg’s syndrome. Int J Pediatr Otorhinolaryngol (2003) 67, 1133-1142.
17. Isabelle Roux, Saaid Safieddine,Regis Nouvian, M’hamed Grati, Marie-Christine Simmler, Amel Bahloul, Isabelle Perfettini, Morgane Le Gall, Philippe Rostaing, Ghislaine Hamard, Antoine Triller, Paul Avan, Tobias Moser, Christine Petit. Otoferlin, Defective in a Human Deafness Form, Is Essential for Exocytosis at the Auditory Ribbon Synapse. Cell (2006) 127, 277–289.
18. Wayne T. Shaia, Steven M. Shapiro, Robert F. Spencer. The Jaundiced Gunn Rat Model of Auditory Neuropathy/Dyssynchrony. Laryngoscope (2005) 115: pp 2167-2173.
19. Steven M. Shapiro. Definition of the Clinical Spectrum of Kernicterus and Bilirubin-Induced Neurologic Dysfunction (BIND). Journal of Perinatology (2005) 25:54–59
20. Perez H, Vilchez J, Sevilla T, Martinez L. Audiologic evaluation in Charcot-Marie-Tooth disease. Scand Audiol Suppl (1988) 30:211-13.
21. Bahr M, Andres F, Timmerman V, Nelis ME, Van Broeckhoven C, Dichgans J. Central visual, acoustic, and motor pathway involvement in a Charcot-Marie-Tooth family with an Asn205Ser mutation in the connexion 32 gene (in process citation). J Neurol Neurosurg Psychiatry (1999) 66:202-6.
22. Butinar D, Zidar J, Leonardis L, Popovic M, Kalaydjieva L, Angelicheva D, Sininger Y, Keats B, Starr A. Hereditary auditory, vestibular, motor, and sensory neuropathy in a Slovenian Roma (Gypsy) kindred. Ann Neurol (1999) 46:36-44.
23. Starr A, Michalewski HJ, Zeng FG, Fujikawa-Brooks S, Linthicum F, Kim CS,
Winnier D, Keats B. Pathology and physiology of auditory neuropathy with a novel
mutation in the MPZ gene (Tyr145->Ser). Brain (2003)126(7):1604-19.
24. Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet (1999) 21:363-9.
25. Varga, R., Kelley, P.M., Keats, B.J., Starr, A., Leal, S.M., Cohn, E., Kimberling, W.J. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J. Med. Genet. (2003) 40, 45–50.
26. Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A (2005) 138:6–10.
27. Sedigheh Delmaghani, Francisco J del Castillo, Vincent Michel, Michel Leibovici, Asadollah Aghaie, Uri Ron, Lut Van Laer, Nir Ben-Tal, Guy Van Camp, Dominique Weil, Francina Langa, Mark Lathrop, Paul Avan, Christine Petit. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nature Genetics (2006) 38 (7): 770-778.
28. L. Stein, K. Tremblay, J. Pasternak, S. Banerjee, K. Lindemann, N. Kraus. Brainstem abnormalities in neonates with normal otoacoustic emissions. Semin. Hear. 17 (2) (1996) 197-212.
29. Rhee Chung-Ku, Park Hyun-Min, Jang Yong-Ju. Audiologic Evaluation of Neonates With Severe Hyperbilirubinemia Using Transiently Evoked Otoacoustic Emissions and Auditory Brainstem Responses. Laryngoscope 109 (1999) 2005-2008.
30. J. Marko, A. Morant, M. Orts, M.I. Pitarch, J. Garcia. Auditory neuropathy in children. Acta Otolaryngol. 120 (2000) 201-204.
31. P. Deltenre, A.L. Mansbach, C. Bozet, F. Christiaens, P. Barthelemy, D. Paulissen, T. Renglet. Auditory neuropathy with preserved cochlear microphonics and secondary loss of otoacoustic emissions. Audiology 38 (1999) 187-195.
32. Starr, A., Sininger, Y. S., Winter, M., Dereby, M. J., Oba, S., Michalewski, H. J. Transient deafness due to temperature-sensitive auditory neuropathy. Ear and Hearing (1998) 19: 169-179.
33. Berlin C, Bordelon J, St John P, D. Wilensky, A. Hurley, E. Kluka, L.J. Hood. Reversing click polarity may uncover auditory neuropathy in infants. Ear Hear (1998) 19:37–47.
34. Zeng FG, Oba S, Sininger Y, Starr A. Temporal and speech processing deficits in auditory neuropathy, Neuroreport 10:3429–3435, 1999.
35. Peterson A, Shallop J, Driscoll C, Breneman A, Babb J, Stoeckel R, Fabry L. Outcomes of cochlear implantation in children with auditory neuropathy. J Am Acad Audiol. (2003) 14(4):188-201.
36. Rosamaria Santarelli, Pietro Scimemi, Erica Dal Monte, Elisabetta Genovese, Edoardo Arslan. Auditory neuropathy in systemic sclerosis: a speech perception and evoked potential study before and after cochlear implantation. Eur Arch Otorhinolaryngol (2006) 263: 809–815.
37. Miyamoto RT, Kirk KI, Renshaw J, Hussain D. Cochlear implantation in auditory neuropathy. Laryngoscope (1999) 109(2 Pt 1):181-5.
38. Joint Committee on Infant Hearing, Position statement: principles and guidelines for early hearing detection and intervention programs, Pediatrics (2000) 6(4): 798-817.