Cubic difference tones (2f1-f2) produced by high-intensity stimuli: their origin revealed by the combined effects of cochlear ischemia and auditory fatigue summarized from: Mom et al. (2001), J.Acoust.Soc.Am. and Avan et al. (2001), Noise and Health.
Thierry Mom, Pierre Bonfils (1), Laurent Gilain, and Paul Avan
Laboratory of Sensory Biophysics (EA 2667), School of Medicine, Clermont-Ferrand, France and (1) Laboratoire de Recherche sur l'Audition, Formation Associ�e Claude-Bernard and Unit� CNRS UPRESA 7060, H�pital Europ�en Georges-Pompidou, Paris, France
e-mail: Paul Avan
1. Introduction
Because otoacoustic emissions (OAE) are thought to arise from the feedback loop allowing outer hair cells (OHC) to enhance the sensitivity and tuning of the organ of Corti, they should vanish whenever a disrupted OHC function results in hearing loss of sufficient degree. This line of reasoning underpins all OAE-based hearing screening procedures. The existence of residual 2f1-f2 cubic difference tones (CDT) during complete cochlear ischemia when stimulus intensities at f1 and f2 exceed 50-60 dB SPL apparently contradicts this tenet.
A first way of overcoming this contradiction consists in denying that residual high-intensity CDTs have anything to do with "active" cochlear mechanisms. "Active / passive" models such as that coined by Mills (1997) assume that CDTs are the vectorial sum of two components, one from the "active" modality, that would be dominant in a healthy cochlea, and one from some "passive" modality that, although normally smaller, would be revealed at high intensities after the "active" component had faded out. The "passive" modality has remained elusive so far: it could arise from entirely "passive" mechanisms per se, such as basilar membrane or inner hair cell motion, or originate from the same nonlinearity as "active" CDTs except that it would come from other places than that tuned to f2. The "active / passive" model has gained popularity by successfully predicting several yet unexplained phenomena, such as the sharp notches occurring in CDT growth functions, or the ones observed along the course of cochlear ischemia or furosemide poisoning: such notches are to be expected whenever the "active" and "passive" components happen to have the same amplitudes and be out of phase. Indeed, the CDT phases (after vs. before the notch) present 180� differences in the case of furosemide (Mills and Rubel, 1994), though not in that of ischemia (Mom et al., 1997, 2001). Besides, Lukashkin and Russell (1999) have designed a single-component model of CDT production that predicts notches despite the absence of a second "passive" modality, by means of letting the operating point of the nonlinear device at the origin of CDTs move with stimulus intensity (which Frank and Koessl had achieved experimentally with similar results in 1996). The argument is more than an academic one because the existence of "passive" CDTs would jeopardize the whole CDT-based screening procedures: most commercial equipments propose default stimulus intensities in excess of 60 dB SPL for the sake of signal-to-noise considerations.
In a first subset of gerbils exposed to complete interruption of their cochlear blood flow, Mom et al. (2001) probed the "active / passive" model. CDT levels and phases were continuously monitored in response to 50 and 60 dB SPL stimuli. After a clear initial decay, CDTs elicited at 60 dB SPL plateaued for several min at about 20 dB below initial level, and could have been "passive" ones; however, whilst sharp level notches were observed in > 50% cases just before the plateau was reached, the attendant CDT phase changes never exceeded 90�. Meanwhile, the CDT group delays decreased by less than 30%. More CDT level notches occurred after more than 10 min following ischemia onset and this time, were associated with sharp phase reversals, however the close similarity between CDT characteristics before and after a notch was hardly consistent with an "active / passive" interpretation. Its failure prompted Mom et al. (2001) to contend that ischemia -or, for this matter, furosemide injection- may not be the best means of knocking CDTs down: they affect OHCs chiefly by decreasing their electrical drive through strial dysfunction whilst OHC stereocilia have no reason to be immediately damaged. Cochlear distortion likely stems from OHC stereocilia bundles as they present well-documented nonlinearities (Flock and Strelioff, 1984; Kros et al., 1992; Lukashkin and Russell, 1999). Accordingly, the next goal of Mom et al. (2001) was, in the first place, to submit gerbil ears to mild pure-tone overexposure so as to induce controlled dysfunction of OHCs and their stereocilia bundles in a narrow interval -obeying the half-octave shift law. Exposure level and duration (90-95 dB SPL, 15-30 min) were such that CDTs remained present even in the frequency range of maximum fatigue. The effect of subsequent ischemia on fatigued CDTs was then compared to that on CDTs from unexposed sites of the cochlea.
2. Methods (in summary)
Anaesthetized adult Mongolian gerbils (3-4 months, 55-75 g).
Auditory bulla opened dorsally. Silver-wire electrode against the round-window membrane, for cochlear-potential monitoring; optic fiber probe (Laser Doppler velocimeter Perimed PF 4000, probe 418, B500-0, 0.5 mm diameter) against the bone of the first cochlear turn; eighth-nerve complex exposed at the porus of the internal auditory meatus; cochlear blood flow interrupted by pressure applied to the porus acousticus by a fire-shaped glass pipette. CDTs monitored by means of the CubeDis Mimosa Acoustics equipment (Ariel DSP 16+, Etymotic earphones and microphone).
- Collection of DPgrams serving as initial references (Figure 1);
- Right ear exposed to a loud pure tone at fexpo (8 kHz in Figure 1) at 90 dB SPL for 15 min; (if insufficient, 95 dB SPL for 15-30 min);
- Post-exposure DPgrams recorded every 5 min until the effects of auditory fatigue on CDTs were stabilized after initial partial recovery (15-30 min);
- Cochlear blood supply blocked, then DPgrams collected every min (60 and 70 dB SPL, f2 = 4 to 16 kHz) for 20 min, then after 20 min, complete series of DPgrams every 5 min for 40 min.
3. Results
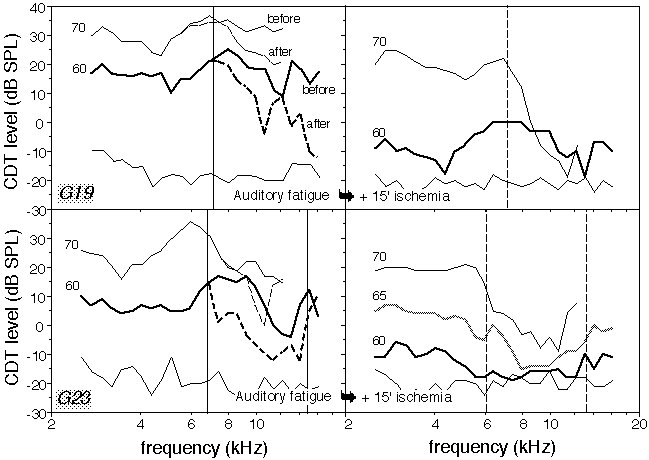
The first DPgrams after loud-tone exposure revealed a decrease in CDT levels at several frequencies f2 > fexpo, relative to the DPgrams before tone exposure. In the ears presenting a V-shaped high-frequency CDT notch, the effect was largest for f2 around half an octave above fexpo. Partial recovery was observed during the first 15 min after the end of exposure, then the CDT levels stabilized so that after 30 min, the largest CDT decrease was 15 to 30 dB for f2 ranging from 10 to 15 kHz. Smaller but significant CDT decreases were present in a frequency interval of about one octave (typical DPgrams after 15 min in Figure 1). The boundaries between unaltered and fatigued ranges were defined on 60-dB-SPL DPgrams, as the smallest and largest frequencies, f2min and f2max, with CDT level changes > -1 dB (see Figure 1, thin vertical lines on left-hand panels; for example, in G23, f2min = 6.5 kHz and f2max = 14.5 kHz).
The behavior of ischemic CDTs in fatigued ears was highly frequency-dependent. In the frequency range where CDTs had been unaffected by exposure to the loud tone, their level and phase changes induced by ischemia were similar to those in unexposed ears, and post-ischemic CDT levels remained comparatively high for 70 dB SPL primaries.
Consider now the range marked with dashed vertical lines (6 < f2 < 13 kHz) in Figure 1 (right-hand panels), that is, f2s with a CDT level more than 5 dB below the average level computed within fatigue-free intervals. It almost coincides with the fatigued area [f2min, f2max], and in this range, CDTs dropped down to within 10 dB of the noise floor whatever the primary intensity up to 75 dB SPL. Figure 1 depicts two similar examples tested up to 70 dB SPL. Note that the fatigue-induced pre-ischemic decrease was too small to account for the large level difference of ischemic CDTs above vs. below 7 kHz. The intervals with dramatically decreased post-ischemic CDTs coincided with the fatigued ones within less than 1.5 kHz in all ears. The dramatic drop of CDTs in fatigued intervals was clearly seen between 3 and more than 30 min after ischemia onset.
Figure 1:(Upper panels) Left side: Series of DPgrams representing an example (in animal G19) of initial CDT levels against frequency f2 at primary intensities of 70 (continuous line) and 60 dB SPL (bold continuous line), and CDT levels 30 min after exposure to a loud tone at fexpo = 8 kHz (primary intensities: see labels next to every plot). The thin vertical line marks the frequency interval outside of which CDT levels at 60 dB SPL were not changed by the tonal exposure. The thin continuous line (bottom) represents the average noise floor. Right side: Same ear 15 min after ischemia onset; The dashed vertical line marks the frequency interval outside of which high-intensity CDTs were smaller than in unexposed ears. (Lower panels) other example from ear G23 (same conventions as for the upper panels). |
4. Discussion
The way mild auditory fatigue modified the subsequent response of CDTs to complete ischemia provides strong arguments against the concept of "passive" ischemic CDTs. The consequences on cochlear micromechanics of auditory fatigue due to mild pure-tone exposure are known to be limited and reversible, and most likely due to a loss of sensitivity of mechano-electrical transduction channels (Patuzzi, 1998). In the present experiment, stimuli with f2 outside the fatigued range generated large high-intensity CDTs -the same as in the absence of preliminary sound exposure. Conversely, when f2 fell in the range where CDT levels had been affected by sound exposure, little or no CDTs were generated even at 70 dB SPL: post-ischemic high-intensity CDTs were therefore highly vulnerable to pre-existing mild auditory fatigue. The most parsimonious interpretation is that they were produced by the same elements that suffered from auditory fatigue. The OHC mechanoelectrical transduction channels being both nonlinear (Flock and Strelioff, 1984; Patuzzi et al., 1989b; Kros et al., 1992; Jaramillo et al., 1993) and sensitive to auditory fatigue (Patuzzi, 1998) are the best candidates for being the sources of CDTs, including the high-intensity ones. Finally, since post-ischemic high-intensity CDTs were affected by auditory fatigue for f2 falling in the range closely obeying the regular tonotopic rules of active mechanisms, they cannot have come from places basal or apical to f2.
These results do not necessarily contradict the widespread idea that the presence of CDTs is strongly correlated with OHC loop function, hence with cochlear sensitivity and tuning. In an ischemic cochlea, the residual OHC feedback loop undoubtedly fails to achieve its main purpose. Even though OHC channels may function -and they do function almost immediately if reperfusion occurs early enough (Mom et al, 1997)-, loop systems are very sensitive to the amount of feedback and it is likely that the ischemia-induced drop in the voltage difference between endolymph and cell led to a dramatic gain decrease. When some cochlear pathology occurs, although BM low-intensity sensitivity and intermediate-intensity compression are largely lost, BM motion catches up with the normal one around 80-90 dB SPL (Sellick et al., 1982; Ruggero and Rich, 1991). We suggest that with intact nonlinear OHC stereocilia and enough BM motion (thus high stimulus intensities), CDTs could still get produced despite ischemia. However, by temporarily damaging OHC stereocilia, auditory fatigue hampered CDT generation (whatever the stimulus intensity) enough to make them vanish.
5. Conclusion
Far from "passive" and robust, residual ischemic CDTs proved to be highly vulnerable to mild auditory fatigue applied prior to ischemia. It strongly suggests that the generation of ischemic CDTs crucially depends on the integrity of mechano-electrical transduction channels. Since the pattern of absence of high-intensity CDTs closely resembled that of auditory fatigue, it is likely that when they existed, high-intensity CDTs were locally produced following the usual tonotopy. A one-component model of CDT generation seems more parsimonious that a two-component one (Mills, 1997) to account for these results. It follows that early stages of cochlear ischemia and presumably euthanasia provide deceptive models of a "passive" cochlea as far as distortion is concerned. This caveat can also be turned as an advantage: persistent high-intensity CDTs despite cochlear dysfunction may mean that the mechano-electrical transduction channels are yet intact -for instance because the dysfunction is of strial origin. If it were confirmed, the protocols using comparatively high primary intensities (Kemp, 1992) would turn out to be particularly useful.
6. Most relevant references
Avan, P., Bonfils, P. and Mom, T. (2001). "Correlations among distortionproduct otoacoustic emissions, thresholds and sensory cell impairments,"Noise & Health 3, 1-17.
Flock, A. and Strelioff D. (1984). "Graded and nonlinear mechanical properties of sensory hairs in the mammalian hearing organ," Nature 310, 597-599.
Frank, G. and Koessl, M. (1996). "The acoustic two-tone distortions 2f1-f2 and f2-f1 and their possible relation to changes in the operating point of the cochlear amplifier," Hear.Res. 98, 104-115.
Jaramillo, F., Markin, V.S. and Hudspeth, A.J. (1993). "Auditory illusions and the single hair cell," Nature 364, 527-529.
Kemp, D.T. (1992). User manual for the ILO92 Otodynamics equipment (Otodynamics, London).
Kros, C.J., Rusch, A. and Richardson, G.P. (1992). "Mechano-electrical transducer currents in hair cells of the cultured neonatal mouse cochlea," Proc.Roy.Soc.Lond.B Biol.Sci. 249, 185-193.
Lukashkin, A.N. and Russell, I.J. (1999). "Analysis of the f2 - f1 and 2f1 - f2 distortion components generated by the hair cell mechanoelectrical transducer: Dependence on the amplitudes of the primaries and feedback gain," J.Acoust.Soc.Am. 106, 2661-2668.
Mills, D.M. (1997). "Interpretation of distortion product otoacoustic emissions measurements: I. two stimulus tones," J.Acoust.Soc.Am. 102, 413-429.
Mills, D.M. and Rubel, E.W. (1994). "Variation of distortion product otoacoustic emissions with furosemide injection," Hear.Res. 77, 183-199.
Mom, T., Avan, P., Romand, R. and Gilain, L. (1997). "Monitoring of functional changes after transient ischemia in gerbil cochlea," Brain Res. 751, 20-30.
Mom, T., Bonfils, P., Gilain, L. and Avan, P. (2001), "Origin of cubicdifference tones generated by high-intensity stimuli: effect of ischemiaand auditory fatigue on the gerbil cochlea," J.Acoust.Soc.Am. 110,1477-1488.
Patuzzi, R.B. (1998). "A four state kinetic model of the temporary threshold shift after loud sound based on inactivation of hair cell transduction channels," Hear.Res. 125, 39-70.
Ruggero, M.A. and Rich, N.C. (1991). "Furosemide alters organ of Corti mechanics: evidence for feedback of outer hair cells upon the basilar membrane," J.Neurosci. 11, 1057-1067.
|